 Cheeta Consumer-GPU Hour-level Exhaustive Epistasis Testing Algorithm
Cheeta Consumer-GPU Hour-level Exhaustive Epistasis Testing Algorithm
Welcome to Cheeta v1.0!
Complete genome-wide SNP×SNP interaction scanning of biobank data within 24 hours.
Introduction
Cheeta v1.0 (C++) has the following main functions.
1. Cheeta is a GPU-accelerated toolkit for exhaustive genome-wide SNP-SNP interaction analysis. It provides three complementary models to accommodate diverse biological hypotheses: the nine-genotype model, the multifactor dimensionality reduction (MDR) model, and the dominant-recessive model. All methods run on a single consumer-grade GPU and are capable of processing biobank-scale datasets.
Pre-installation
Cheeta v1.0 is implemented by C++. Before using it, please install CUDA programing environment (CUDA 12 or update) first.
Download
- Download Cheeta v1.0 (C++, Command-line running, windows) a GPU-accelerated parallel computing software for Windows.
Nine-Genotype Model (genotype_interaction)
This model exhaustively evaluates all nine possible joint genotype combinations of two SNPs. For each combination, a 2×2 contingency table is constructed and tested for association with case/control status.
Usage
command-line example:
cheeta genotype_interaction -file0 -file1 -o [-threads ] [-alpha_cut ] [-or_cut ] [-set_gpu ]
parameters
cheeta: Perform genome-wide interaction analysis
genotype_interaction: exhaustively evaluates all nine possible joint genotype combinations of two SNPs.
-file0: Path to the case genotype file. Rows: SNPs, columns: individuals. Values: 0 (AA), 1 (Aa), 2 (aa), 9 (missing). Tab-separated.;
-file1: Path to the control genotype file. Same format as -file0.;
-o: Output file path.;
-alpha_cut: Significance threshold for the chisquare-value.(default: 1e-5);
-or_cut: Odds ratio confidence interval filter. Only pairs whose OR confidence interval lies entirely outside [1/θ, θ] are reported (default: 2.0);
-threads: Number of threads per GPU block. Usually left at default.(default:256);
-set_gpu: GPU device ID to use (useful for multi‑GPU systems).(default:0);
Input/Output Format
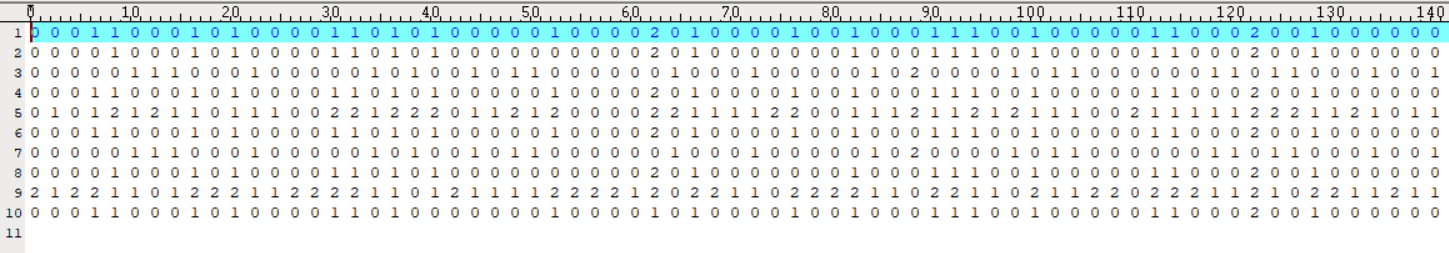
Input File: please see "case.txt" and "control.txt" for more information.
- case.txt:
- control.txt:
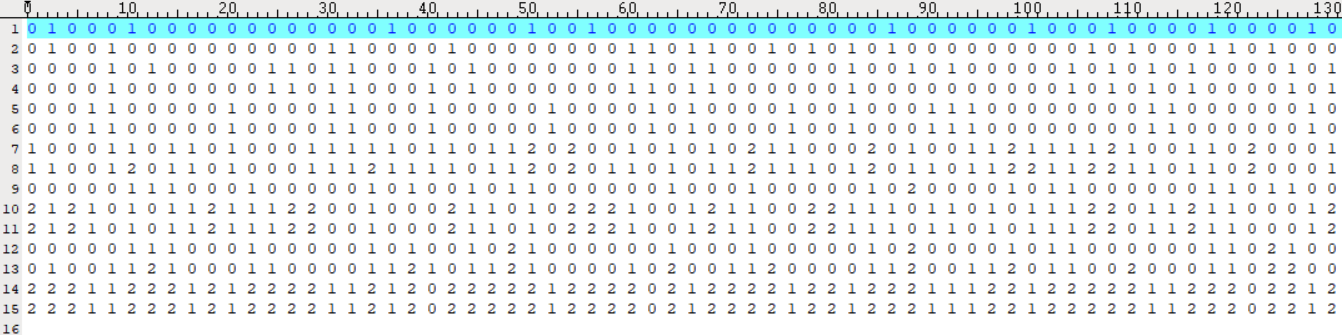
Both input files share the same format: each colomn represents a sample, and each row corresponds to the genotype of an SNP (coded as 0, 1, or 2, representing genotypes AA, Aa, and aa, respectively). The input files must be pre-processed according to the reference genome prior to analysis.
Output File: please see output.txt for more information.
Genotype_Label SNP0 SNP1 a b c d chi_square chi_pvalue OR OR_lower OR_upper
Genotype_Label: One of the nine combinations, e.g., AA*BB_vs_other.
SNP0, SNP1: Zero-based indices of the two SNPs in the input file.
a: Case count with the target genotype combination.
b: Case count with any other combination.
c: Control count with the target combination.
d: Control count with any other combination.
chi_square: Yates-corrected Chisq-square statistic.
chi_pvalue: P-value from the Chisq-square test.
OR: Point estimate of the odds ratio.
OR_lower, OR_upper: 95% confidence interval of the OR.
Example
cheeta genotype_interaction -file0 cases.txt -file1 controls.txt -o nine_geno_result.txt -alpha_cut 1e-6 -or_cut 2.0
Multifactor Dimensionality Reduction (MDR) Model (mdr_interaction)
This model collapses the nine genotype combinations into two risk categories (high‑risk vs. low‑risk) using a sample‑size correction factor, then performs a single statistical test per SNP pair.
Usage
command-line example:
cheeta mdr_interaction -file0 -file1 -o [-threads ] [-alpha_cut ] [-or_cut ] [-set_gpu ]
parameters
cheeta: Perform genome-wide interaction analysis
mdr_interaction: exhaustively evaluates all high/low risk genotype combinations of two SNPs.
-file0: Path to the case genotype file. Rows: SNPs, columns: individuals. Values: 0 (AA), 1 (Aa), 2 (aa), 9 (missing). Tab-separated.;
-file1: Path to the control genotype file. Same format as -file0.;
-o: Output file path.;
-alpha_cut: Significance threshold for the chisquare-value.(default: 1e-5);
-or_cut: Odds ratio confidence interval filter. Only pairs whose OR confidence interval lies entirely outside [1/θ, θ] are reported (default: 2.0);
-threads: Number of threads per GPU block. Usually left at default.(default:256);
-set_gpu: GPU device ID to use (useful for multi‑GPU systems).(default:0);
Input/Output Format
Input File: please see "case.txt" and "control.txt" for more information.
- case.txt:
- control.txt:
Both input files share the same format: each column represents a sample, and each row corresponds to the genotype of an SNP (coded as 0, 1, or 2, representing genotypes AA, Aa, and aa, respectively). The input files must be pre-processed according to the reference genome prior to analysis.
Output File: please see output.txt for more information.
Model SNP0 SNP1 a b c d chi_square chi_pvalue OR OR_lower OR_upper
Model: Always high_risk_vs_low_risk.
SNP0, SNP1: Zero-based indices of the two SNPs in the input file.
a: Case count with the target genotype combination.
b: Case count with any other combination.
c: Control count with the target combination.
d: Control count with any other combination.
chi_square: Yates-corrected Chisq-square statistic.
chi_pvalue: P-value from the Chisq-square test.
OR: Point estimate of the odds ratio.
OR_lower, OR_upper: 95% confidence interval of the OR.
Example
cheeta mdr_interaction -file0 cases.txt -file1 controls.txt -o mdr_result.txt -alpha_cut 1e-5 -or_cut 1.5
Dominant‑Recessive Model (domrec_interaction)
This model implements four classical Mendelian inheritance patterns based on the reference alleles: Dominant‑Dominant (DD), Dominant‑Recessive (DR), Recessive‑Dominant (RD), and Recessive‑Recessive (RR). For each pattern, a single 2×2 table is tested per SNP pair.
Usage
command-line example:
cheeta domrec_interaction -file0 -file1 -o [-threads ] [-alpha_cut ] [-or_cut ] [-set_gpu ]
parameters
cheeta: Perform genome-wide interaction analysis
domrec_interaction: exhaustively evaluates all dominant/recessive genotype combinations of two SNPs.
-file0: Path to the case genotype file. Rows: SNPs, columns: individuals. Values: 0 (AA), 1 (Aa), 2 (aa), 9 (missing). Tab-separated.;
-file1: Path to the control genotype file. Same format as -file0.;
-o: Output file path.;
-alpha_cut: Significance threshold for the chisquare-value.(default: 1e-5);
-or_cut: Odds ratio confidence interval filter. Only pairs whose OR confidence interval lies entirely outside [1/θ, θ] are reported (default: 2.0);
-threads: Number of threads per GPU block. Usually left at default.(default:256);
-set_gpu: GPU device ID to use (useful for multi‑GPU systems).(default:0);
Input/Output Format
Input File: please see "case.txt" and "control.txt" for more information.
- case.txt:
- control.txt:
Both input files share the same format: each column represents a sample, and each row corresponds to the genotype of an SNP (coded as 0, 1, or 2, representing genotypes AA, Aa, and aa, respectively). The input files must be pre-processed according to the reference genome prior to analysis.
Output File: please see output.txt for more information.
Model SNP0 SNP1 a b c d chi_square chi_pvalue OR OR_lower OR_upper
Model: One of the four patterns, e.g., (AA+Aa)*(BB+Bb)_vs_other (DD).
SNP0, SNP1: Zero-based indices of the two SNPs in the input file.
a: Case count with the target genotype combination.
b: Case count with any other combination.
c: Control count with the target combination.
d: Control count with any other combination.
chi_square: Yates-corrected Chisq-square statistic.
chi_pvalue: P-value from the Chisq-square test.
OR: Point estimate of the odds ratio.
OR_lower, OR_upper: 95% confidence interval of the OR.
Example
cheeta domrec_interaction -file0 cases.txt -file1 controls.txt -o domrec_result.txt -alpha_cut 1e-5 -or_cut 2.0